DeltaSCF-CASTEP-Module¶
This document explains the use of the deltascf.f90 module in
CASTEP version 17.1. This module has been developed at the Technische Universität München.
This module allows to calculate excited states via 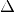 SCF calculations of different
kinds as well as to apply penalty potentials to reference
orbitals. It also allows to calculate the projected DOS onto these
reference orbitals with the post-processor MolPDOS.
SCF calculations of different
kinds as well as to apply penalty potentials to reference
orbitals. It also allows to calculate the projected DOS onto these
reference orbitals with the post-processor MolPDOS.
The corresponding references are:
- for le SCF and MolPDOS: R. J. Maurer and K. Reuter, J. Chem. Phys. 139, 014708 (2013)
- for DFT+U(MO): M. Müller, K. Diller, R. J. Maurer, and K. Reuter, J. Chem. Phys. 144, 024701 (2016)
- for SCF-DFT see also: R. J. Maurer, and K. Reuter, J. Chem. Phys. 135, 224303 (2011)
or
First-Principles Description of the Isomerization Dynamics of Surface-Adsorbed Molecular Switches, Doctoral Thesis, Technische Universität München, 2014
Here is a list of publications, which have employed functionality from this module:
- Maurer, K. Reuter, “Bistability Loss as a Key Feature in Azobenzene (Non-) Switching on Metal Surfaces”, Angew. Chem. Int. Ed. 51, 12009-12011 (2012)
- Gopakumar, T. Davran-Candan et al., “Broken Symmetry of an Adsorbed Molecular Switch Determined by Scanning Tunneling Spectroscopy”, Angew. Chem. Int. Ed. 52, 11007-11010, (2013)
- Scheil, T. G. Gopakumar, et al., “Switching of an Azobenzene-Tripod Molecule on Ag(111)”, J. Phys. Chem. Lett. 7, 2080-2084 (2016)
- Karan, N. Li, et al., “Spin Manipulation by Creation of Single-Molecule Radical Cations”, Phys. Rev. Lett. 116, 027201 (2016)
- Müller, K. Diller, R. J. Maurer, K. Reuter, “Interfacial charge rearrangement and intermolecular interactions: Density-functional theory study of free-base porphine adsorbed on Ag(111) and Cu(111)”, J. Chem. Phys. 144, 024701, (2016)
For more information, please contact reinhard.maurer@yale.edu