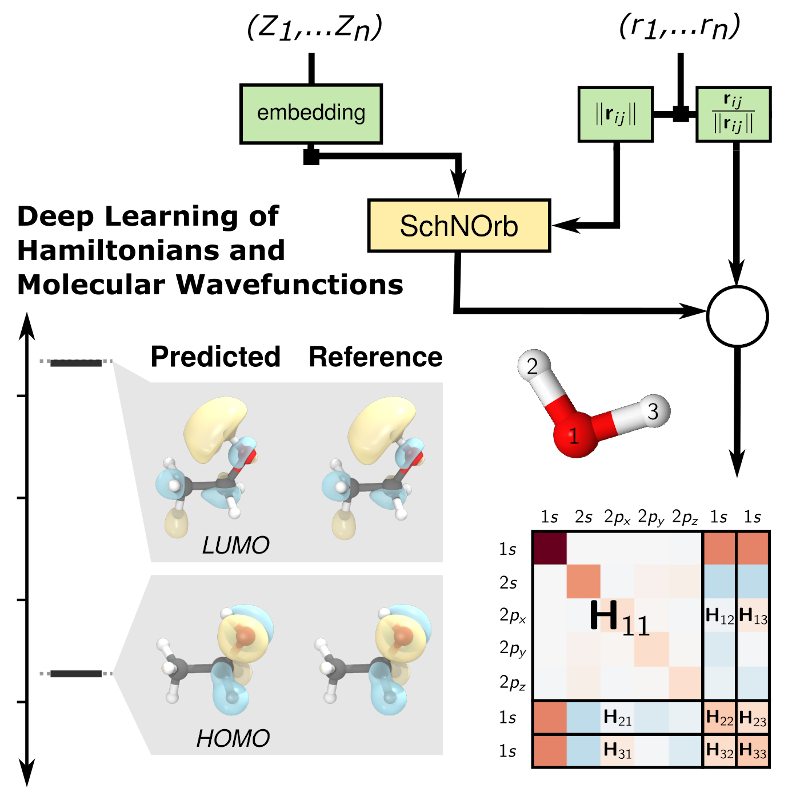
With the recent emergence of machine-learning-based potential energy interpolation, it is clear that machine learning (ML) must play an important role in bridging between atomistic and experimentally-accessible time and length scales. The prediction of electronic and spectroscopic properties and chemical reactivity is heavily reliant on non-scalar quantum mechanical observables beyond the potential energy such as time-dependent response functions. In our work, we us convolutional continuous layer neural networks to predict non-scalar quantum mechanical properties such as electron-phonon response functions and the quantum mechanical molecular Hamiltonian of molecules and materials as a function of all atomic positions. Our goal is to develop highly accurate and efficient ML-enhanced tight-binding-based simulation methods that provide access to response properties at unprecedented time and length scales based on first principles data generated by Density Functional Theory.
Current Projects
- Machine-learning parametrisation of lattice-site Hamiltonians to accelerate mixed quantum-classical dynamics simulations
- Machine learning representation of Kohn-Sham Hamiltonians and other fundamental electronic structure properties
- Integration of machine learning surrogate models and electronic structure software
- Machine learning surrogates of electron-phonon coupling and electronic friction
Collaborations
- Prof. James Kermode, University of Warwick, UK
- Prof. Christoph Ortner, University of British Columbia, UK
- Dr. Matthias Sachs, Lancaster University, UK